
Design of leucine-rich repeat kinase 2 (LRRK2) inhibitors using a crystallographic surrogate derived from checkpoint kinase 1 (Chk1)
Williamson et al., J. Med. Chem. 2017
Mutations in leucine-rich repeat kinase 2 (LRRK2), such as G2019S, are associated with an increased risk of developing Parkinson’s disease. Surrogates for the LRRK2 kinase domain based on checkpoint kinase 1 (Chk1) mutants were designed, expressed in insect cells infected with baculovirus, purified, and crystallized. X-ray structures of the surrogates complexed with known LRRK2 inhibitors rationalized compound potency and selectivity. The Chk1 10-point mutant was preferred, following assessment of surrogate binding affinity with LRRK2 inhibitors.
Fragment hit-derived arylpyrrolo[2,3-b]pyridine LRRK2 inhibitors underwent structure-guided optimization using this crystallographic surrogate. LRRK2-pSer935 HEK293 IC50 data for 1 were consistent with binding to Ala2016 in LRRK2 (equivalent to Ala147 in Chk1 10-point mutant structure). Compound 1 was shown to be potent, moderately selective, orally available, and brain-penetrant in wild-type mice, and confirmation of target engagement was demonstrated, with LRRK2-pSer935 IC50 values for 1 in mouse brain and kidney being 1.3 and 5 nM, respectively. Read more
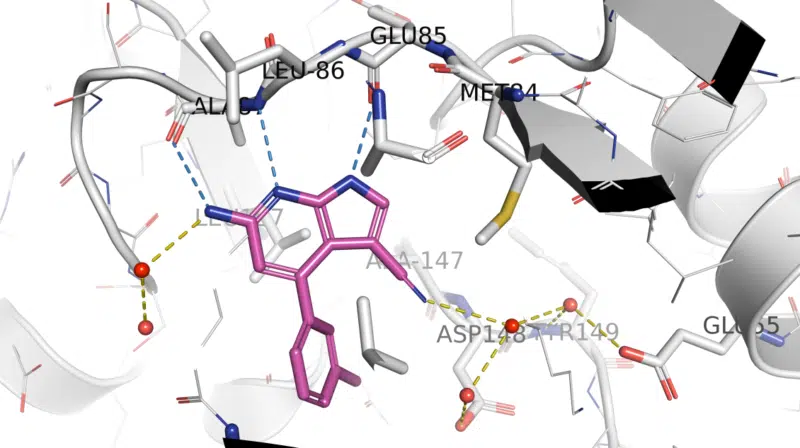
X-ray crystal structure of 1/ Chk1 10-pt. mut. (PDB: 5OPU)